¡Descarga Reumatología articulos y más Guías, Proyectos, Investigaciones en PDF de Reumatología solo en Docsity!
Horizon scan: State-of-the-art therapeutics for
psoriatic arthritis
Joseph Hutton
a, *
, Philip Mease
b
, Deepak Jadon
c
a (^) Department of Rheumatology, Cambridge University Hospitals NHSFT, Hills Road, Cambridge, CB2 0QQ, UK b (^) Seattle Rheumatology Associates, 601 Broadway, Suite 600, Seattle, WA 98122, USA c (^) Rheumatology Research Unit, Department of Medicine, University of Cambridge, Cambridge, CB2 0QQ, UK
Keywords: Psoriatic arthritis (PsA) Spondylarthritis Therapeutics IL IL12/ Janus kinases Tyrosine kinase 2 Mitogen activated protein kinase 2 Nano-IL-17 inhibitors Nanobodies
a b s t r a c t
Psoriatic arthritis (PsA) is a common immune-mediated inflam- matory disease (IMID) that can present with a heterogenous clinical phenotype. The advent of advanced therapies has sub- stantially improved patient outcomes, but many patients still have suboptimal or unsustained response, resulting in morbidity, structural damage and functional impairment. There remains a need for better therapeutic options and precision medicine ap- proaches to improve outcomes for patients with PsA. This review synthesises recently approved the state-of-the-art therapeutics for PsA, including inhibitors of IL-23, Janus kinase (JAK), tyrosine ki- nase 2 (TYK2) and dual-target IL-17A/F. The evidence base for emerging therapeutics, including MK-2 inhibitors, nano-IL-17 in- hibitors, nanobodies and other dual-target therapies for PsA is also reviewed. Potential future therapeutic strategies and unmet research needs are discussed. © 2022 The Author(s). Published by Elsevier Ltd. This is an open access article under the CC BY license (http://creativecommons. org/licenses/by/4.0/).
Introduction
Psoriatic arthritis (PsA) is a heterogenous immune-mediated inflammatory disease (IMID) that can manifest as enthesitis, dactylitis, synovitis, spondylitis, sacroiliitis, osteoproliferation, osteitis, skin psoriasis, nail psoriasis, uveitis and inflammatory bowel disease (IBD) [1]. It is associated with multiple comorbidities including the metabolic syndrome, obesity, type 2 diabetes mellitus, cardiovascular
- Corresponding author. E-mail addresses: jh2164@cam.ac.uk (J. Hutton), pmease@philipmease.com (P. Mease), dj351@cam.ac.uk (D. Jadon).
Contents lists available at ScienceDirect
Best Practice & Research Clinical
Rheumatology
j o u r n a l h o m e p a g e : w w w. e l s e v i e r h e a l t h. c o m / b e r h
https://doi.org/10.1016/j.berh.2022. 1521-6942/© 2022 The Author(s). Published by Elsevier Ltd. This is an open access article under the CC BY license (http:// creativecommons.org/licenses/by/4.0/).
Best Practice & Research Clinical Rheumatology 36 (2022) 101809
disease, depression, fibromyalgia, non-alcoholic fatty liver disease and increased mortality [2,3]. This can have an impact on a patient's quality of life, functional ability and poses considerable personal and societal burden through work presenteeism and absenteeism, and both direct and indirect healthcare costs [2,4,5]. Cutaneous psoriasis affects approximately 2e3% of the Western population [6], and 20e30% of these patients will develop musculoskeletal disease [2]. Treat-to-target, the principle of therapeutic escalation to achieve a pre-specified objective level of disease activity, has been demonstrated to be beneficial in early stage PsA [7]. Target outcomes might include minimal disease activity (MDA), ACR responses, psoriasis area and severity index (PASI), patient-reported outcome measures (PROMs) and quality of life indices. Longer term benefits remain under investigation [8]. Despite the importance of early diagnosis and treatment, many patients have delayed diagnosis with resultant poorer outcomes [9]. This is in part due to the lack of validated laboratory or imaging biomarkers specific to PsA [10], which can result in underdiagnosis as osteoarthritis (OA) or chronic pain syndromes, or over diagnosis of PsA in other patients. Both have an impact on early and correct diagnosis, as well as treatment response in both clinical practice and trial settings. Despite several therapeutic options being available for PsA (Table 1), including non-steroidal anti- inflammatory drugs (NSAIDs), corticosteroids, conventional synthetic disease-modifying antirheu- matic drugs (csDMARDs), biological DMARDs (bDMARDs) and targeted synthetic DMARDs (tsDMARDs), no treatment consistently achieves an ACR20 response in over 60% of patients [2,11], and approximately 50% of patients have radiographic damage within 2 years of onset [12]. Treatments can fail for several reasons, including primary or secondary inefficacy, adverse effect, poor patient adherence, formation of antidrug antibodies and environmental factors such as obesity, smoking, fatty liver disease and other comorbidities (Fig. 1). Treatments also demonstrate variable efficacy across different disease domains. Efficacy is frequently not achieved (primary inefficacy) or lost (secondary inefficacy; poor persistence) in many patients, and therefore prompt a switch of treatment [2,13]. The differing efficacy across domains emphasises the importance of careful therapy choice and a multispecialty approach to ensure that the patient's wishes, related conditions, comorbidities, and active domains are holistically addressed [14,15]. Many treatments also require close monitoring to ensure patient safety and optimal dosing. Treatment adherence can be negatively impacted by actual and perceived adverse effects from treatment. The inconvenience of frequent blood monitoring and low alcohol consumption required by some csDMARDs also impacts adherence [16].
Table 1 Current therapeutic groups for PsA. Table details the currently approved therapeutic classes and agents for PsA. PsA, psoriatic arthritis.
Adjunctive Therapies Non-steroidal anti-inflammatory drugs (NSAIDs) Glucocorticoids: oral, intra-articular, intra-muscular, topical routes Conventional Synthetic DMARDs (csDMARDs) Methotrexate Leflunomide Sulfasalazine Cyclosporine Biological DMARDs (bDMARDs) TNF Inhibitors: etanercept a^ , infliximab a^ , adalimumab a^ , golimumab, certolizumab IL-12/IL-23 p40 Subunit Inhibitors: ustekinumab IL-23 p19 Subunit Inhibitors: guselkumab, risankizumab b, tildrakizumab b IL-17 Inhibitors: secukinumab, ixekizumab, brodalumabb T-cell Modulator: abatacept Targeted Synthetic DMARDs PDE4 Inhibitor: apremilast JAK Inhibitors: tofacitinib, upadacitinib a (^) Available as biosimilars at the time of writing. b (^) Approved for cutaneous psoriasis but not PsA at the time of writing.
Secondary non-response to treatment is also common. Up to 30% of patients discontinue tumour necrosis factor inhibitors (TNFi) by 1 year, and median survival on initial TNFi is 2.2 years, with drug persistence decreasing further for subsequent agents [25e27]. Drug persistence data suggests that the persistence of tofacitinib and infliximab is the lowest for the drugs used in PsA, with general equiv- alency amongst other drugs [28]. Exceptions to this were golimumab, secukinumab and ixekizumab, which had a slightly higher drug persistence [28]. Biologic therapies are intrinsically immunogenic, and patients with PsA can develop antidrug antibodies at similar rates to other rheumatic diseases [28]. These can decrease serum concentrations to subtherapeutic levels and thus reduce their efficacy [29]. In RA and IBD, the development of anti-drug antibodies has been shown to be attenuated by the concomitant use of csDMARDs such as methotrexate, azathioprine and perhaps leflunomide [29]. The same has not been shown to date when methotrexate was combined with etanercept for PsA [30] or in other RCTs. After initial advanced treatment failure, it is common clinical practice and within international recommendations to switch within the same class (especially for secondary failure) or to a different class (especially for primary failure). However, a recent study challenged this practice, by demon- strating that the outcomes following switching to a different class were no different to those after switching within the same class [31]. There is an urgent need for new therapeutics to expand our clinical armamentarium and address these unmet clinical needs. This review aims to highlight both recently approved and emerging treatments for patients with PsA.
Recently approved therapeutics
IL-17A inhibitors
The interleukin (IL)-17 superfamily consists of 6 different isoforms from IL-17A to IL-17F [32,33]. Of these, IL-17A and IL-17F are the best studied and can exist as either homodimers or heterodimers [32].
IL-17 is primarily produced by T-helper 17 (Th17) and gd T-cells amongst others, particularly in
response to IL-12, IL-23 and IL-1b secreted from monocytes, macrophages and dendritic cells (DCs) on
the engagement of pattern recognition receptors (PRR) [1,32]. PRRs act via IL-17 receptors (IL-17R) that consist of 5 main subtypes, including IL-17 receptor subunits (IL-17R) A and IL-17RC. A range of downstream adaptors including ACT1, tumour necrosis factor receptor associated factors (TRAF)2, 5
and 6 activate the nuclear factor kb (NF-kb) pathway, stabilise messenger RNA (mRNA) and induce pro-
inflammatory mediator production [32,33]. IL-17A is the major target for both secukinumab and ixekizumab (Fig. 2). Uniquely, both ixekizumab and secukinumab have data evaluating their head-to-head performance against the TNFi adalimumab in PsA. A randomised open-label assessor-blinded trial compared ixekizumab with adalimumab in 556 patients with active PsA (bio-naïve, csDMARD-inadequate responders (IR)) [34]. Ixekizumab was statistically more likely than adalimumab at week 24 to achieve the primary hybrid endpoint of simultaneous ACR50 and PASI 100 response (p ¼ 0.036) [34]. No differences in ACR50 response alone were seen between treatment arms (treatment difference 3.9%, 95% CI -4.3%e12.1%), though ixeki- zumab was statistically more likely to achieve a PASI 100 response (p ¼ 0.001). Of note, more Ixekizumab-treated patients achieved the secondary endpoints of MDA (treatment difference 12.4%, 95% CI 4.3e20.4%), very low disease activity (VLDA) (7.1%, 95% CI 1.4e12.7%), PsA disease activity score (PASDAS) defined remission (9.5%, 95% CI 2.5e16.6%) and spondyloarthritis research consortium of Canada (SPARCC) enthesitis index ¼ 0 (11.6%, 95% CI 1.3e21.9%) [34]. No significant differences were observed between treatment groups for disease activity in psoriatic arthritis (DAPSA) change from baseline, DAPSA low disease activity, ACR20, ACR70 and nail psoriasis severity index (NAPSI). Adverse effects were more common in the ixekizumab arm, but most being mild-moderate. Discontinuations due to adverse effects were more common in the adalimumab arm [34]. The EXCEED study compared secukinumab with adalimumab in 853 patients with active PsA (bDMARDs-naïve, csDMARDs-IR) [35]. No statistically significant differences were found between the two agents for reaching the primary endpoint of ACR20 response at week 52 (OR 1.30, 95% CI 0.98e1.72) [35]. As a difference in the primary
endpoint was not met, key secondary outcomes were not tested for statistical significance. Pre- specified exploratory analyses also suggested that more secukinumab patients achieved combined musculoskeletal and skin responses (ACR50 þ PASI 100, OR 1.85, 95% CI 1.17e2.92), skin responses (PASI 75 OR 2.33 95% CI 1.5e3.6, PASI 100 OR 2.01 95% CI 1.34e3.03%, absolute PASI <3 OR 2.
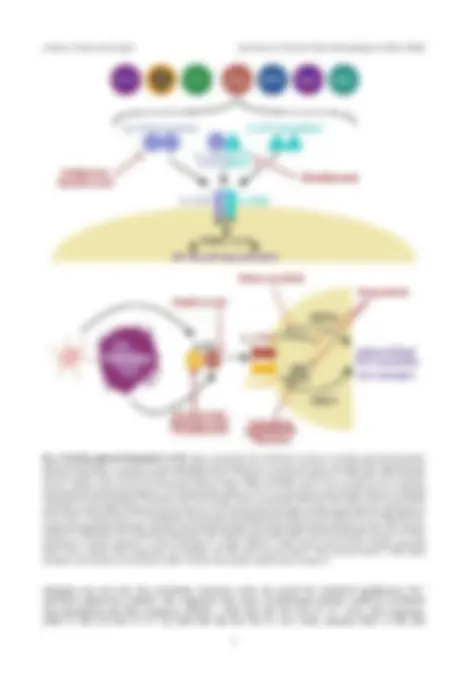
Fig. 2. Recently approved therapeutics in PsA. Figure summarises the mechanism of action of recently approved therapeutic agents for PsA. Briefly, IL-17A and IL-17F are produced by cells including Th17, gd T-cells, ILCs, iNK T-cells, MAIT cells, neutrophils and mast cells as both homo- and hetero-dimers. This binds to the IL-17 receptor consisting of 2 subunits, IL-17RA and IL-17RC, recruiting the ACT1 adaptor, which recruits the downstream effectors TRAF2, TRAF5 and TRAF6, which in turn activate the NF-kb pathway. Ixekizumab and secukinumab inhibits IL-17A. Bimekizumab inhibits IL-17F. IL-23 (consisting of p19 and p40) subunits is produced mainly by DCs and macrophages. This binds to the IL-23 receptor, which recruits JAK1, JAK3 and TYK2, which in turn recruit STAT and STAT4 to induce RORgT production and promote Th17 cell survival and cell activation. Ustekinumab inhibits the p40 subunit of IL-23, which is shared with IL-12. Guselkumab, risankizumab and tildrakizumab inhibit the p19 subunit of IL-23. Filgotinib, tofa- citinib and upadacitinib inhibit JAK1 and JAK3. Deucravacitinib inhibits TYK2. Brepocitinib inhibits both JAK and TYK2. PsA, psoriatic arthritis; IL, interleukin; ILCs, innate-like lymphocytes; iNK, induced natural killer; MAIT, mucosal associated invariant T; IL-17RA, interleukin-17 receptor subunit A; IL-17RC, interleukin-17 receptor subunit C; TRAF, tumour necrosis factor receptor associated factor; NF-kb, nuclear factor kappa beta; DC, dendritic cell; JAK, janus tyrosine kinase; TYK2, tyrosine kinase 2; STAT, signal transducer and activator of transcription; RORgT, retinoic acid receptor related orphan receptor C.
safety are expected in 2026 [44]. Bimekizumab has been approved for the treatment of psoriasis in numerous countries; PsA and axial spondyloarthritis approvals are pending. The drug has not yet been approved by the FDA in the US for various indications.
IL-17RA inhibitors
Brodalumab is a human monoclonal antibody that binds with high affinity to the IL-17RA receptor and thus blocks the action of multiple members of the IL-17 family (Fig. 2) [45]. In two phase III studies, AMVISION-1 and AMVISION-2 assessed the safety and efficacy of brodalumab versus placebo in 962 patients with active PsA (csDMARDs-IR) [45]. Both studies were terminated early due to suicidal ideation and behaviour in treated patients, and thus concerns that this would lead to restrictive labelling [45]. Given the concerns about suicidal ideation, protocol amendment to exclude patients with a prior history was instituted. The overall frequencies of emergent suicidal ideation and behaviour were similar between treated and placebo, suggesting that this did not increase the risk in patients without prior history [45]. Brodalumab is also approved for the treatment of plaque psoriasis in multiple countries worldwide, but, currently, no further trials in PsA are planned at the time of writing [45].
IL-23 inhibitors
IL-23 is a key dimeric cytokine consisting of p19 and p40 subunits and produced mainly by mac- rophages and DCs in PsA [1]. The p40 subunit is also shared with IL-12. IL-23 binds to the IL-23 receptor (IL-23R) and leads to the recruitment of Janus Kinase (JAK)2 and Tyrosine Kinase 2 (TYK2). This induces
the RAR-related orphan receptor gamma (RORgT) production to promote the differentiation and sur-
vival of Th17 cells, as well as the granulocyte-macrophage colony stimulating factor (GM-CSF) pro- duction [1,46]. Ustekinumab targets the shared p40 subunit and therefore antagonises both IL-23 and IL-12 cytokines. More targeted p19 inhibitors have since been developed, that only antagonise IL-23, and are discussed in detail here.
Guselkumab Guselkumab is a novel human monoclonal antibody that targets the p19 subunit of IL-23 [47]. Two phase III trials have assessed its efficacy in PsA, including biologic naïve [48] and TNFi inadequate response patients [47]. The DISCOVER-1 study compared guselkumab and placebo in 381 PsA patients (csDMARDs-IR and bDMARDs-IR), including 30% of patients who had received 1e2 TNFi [47]. Patients treated with guselkumab were statistically more likely than placebo to achieve the primary endpoint of ACR20 response at 24 weeks (100 mg 4 times weekly 59%, 100 mg 8 times weekly 52%, placebo 22%; p < 0.001), including those who had received prior DMARDs [47]. Guselkumab-treated patients were also significantly more likely to achieve secondary endpoints for musculoskeletal outcomes such as ACR50 (100 mg 4 times weekly 36%, 100 mg 8 times weekly 30%, placebo 9%; p < 0.001) and ACR (100 mg 4 times weekly 20%, p ¼ 0.005; 100 mg 8 times weekly 12%, p ¼ 0.069; placebo 6%) [47]. Similar findings were seen for PASI I00 (4 times weekly difference vs. placebo 39%, 95% CI 28e50%, p < 0.001; 8 times weekly 20%, 95% CI 10e30%, p ¼ 0.012) [47]. The DISCOVER-2 study compared 4 times weekly vs. 8 times weekly guselkumab and placebo in 741 patients with active PsA (bDMARDs-naïve) [48]. Significantly greater numbers of patients in the guselkumab arms achieved the primary outcome of ACR20 response at week 24 (4 times weekly difference vs. placebo 31%, 95% CI 22e39; 8 times weekly difference vs. placebo 31%, 95% CI 23e40%; p < 0.0001). Improvements in enthesitis (100 mg 4 times weekly 48%, placebo 27%; p ¼ 0.013) were seen in pooled results [47,48]. Both studies demonstrated similar safety profiles and were comparable to prior reports and other medications. Adverse effects of URTI, nasopharyngitis, bronchitis and raised liver enzymes were observed [47,48].
Risankizumab Risankizumab is a humanised IgG1 antibody against the IL-23 p19 subunit, initially approved for moderate-to-severe plaque psoriasis [49]. Two phase III global multicentre double-blinded placebo-
controlled trials have been performed in PsA [49,50]. KEEPsAKE-1 compared 150 mg risankizumab against placebo at weeks 0, 4 and 16 in 964 patients with active PsA (csDMARD-IR) [49]. Risankizumab treatment was significantly more likely than placebo to achieve the primary endpoint of ACR20 at week 24 (risankizumab 57.3% vs. 33.5% placebo, p < 0.001) [49]. Risankizumab treatment was also signifi- cantly associated with the secondary endpoints of improvements in enthesitis (51.2% vs. 37.2%, p < 0.001), dactylitis (66.95 vs. 54.4%, p ¼ 0.034) [49], skin improvements in those with >3% BSA at baseline (PASI90 52.3% vs. 9.9%, p < 0.001) and nail outcomes (mNAPSI, p < 0.001) [49]. Improvements in physical functioning as measured by significantly greater decreases from baseline in HAQ-QI (p < 0.001), SF-36 (p < 0.001) and FACIT-Fatigue (p < 0.001) were seen with risankizumab versus placebo [49]. KEEPsAKE-2 compared 150 mg risankizumab at weeks 0, 4 and 16 against placebo in 444 patients with active PsA (bDMARDs-IR 46.5%, csDMARDs-IR; but previous IL-23i, IL-12/23i and IL-17i were not permitted) [50]. Risankizumab was significantly more likely to achieve the primary endpoint of ACR20 at week 24 than placebo (51.3% vs. 26.5%, p < 0.001). Significantly higher ACR responses were observed with risankizumab, regardless of whether the patient received concomitant csDMARDs (50.4% vs. 33.9%), risankizumab as monotherapy (53.0% vs. 16%), were csDMARDs-IR (56.3% vs. 36.6%) or were bDMARDs-IR (45.7% vs. 14.9%) [50]. Statistically greater improvements in enthesitis (42.9% vs. 30.4% p < 0.01), dactylitis (72.5% vs. 42.1% p < 0.001), skin (PASI90 55% vs. 10.2% p < 0.001), HAQ-DI (�0.22 vs. �0.05, p < 0.001), SF-36 (5.9 vs. 2.0 p < 0.001) and FACIT-fatigue (25.6% vs. 11.4% p < 0.001) were seen with risankizumab vs. placebo treated patients [50]. Side effects were comparable to other agents and common side effects included nasopharyngitis, URTI, headache and altered liver enzymes [49,50].
Tildrakizumab Tildrakizumab is a high affinity antibody for the p19 subunit of IL-23. Phase IIB studies compared 200 mg tildrakizumab 4 times weekly vs. tildrakizumab 20 mg, 100 mg or 200 mg 12 times weekly vs. 4 times weekly placebo in 391 patients with active PsA (csDMARD-IR, TNFi-IR to a maximum of 30% of the total recruited) [51]. Tildrakizumab was statistically more likely than placebo at week 24 to achieve the primary endpoint of ACR20 response (4 times weekly 200 mg tildrakizumab response rate 79.5 ± 4.6% standard error p ¼ 0.0001, 12 times weekly 200 mg tildrakizumab 77.2 ± 4.7% p ¼ 0.0006, 12 times weekly 100 mg tildrakizumab 71.4 ± 5.2% p ¼ 0.0088, 12 times weekly 20 mg tildrakizumab 73.1 ± 5.0% p ¼ 0.0041, placebo 50.6 ± 5.6%) [51]. Patients receiving 200 mg 4 times weekly or 12 times weekly also achieved higher rates of musculoskeletal improvement compared to placebo, including: ACR50 (4 times weekly 200 mg tildrakizumab 52.6 ± 5.7% p ¼ 0.0002, 12 times weekly 200 mg tildrakizumab 50.6 ± 5.6% p ¼ 0.0006, placebo 24.1 ± 4.8%), ACR70 (4 times weekly 200 mg tildrakizumab 28.2 ± 5.1% p ¼ 0.0040, 12 times weekly 200 mg tildrakizumab 29.1 ± 5.1% p ¼ 0.0033, placebo 10.1 ± 3.4%), DAS28-CRP<3.2 (4 times weekly 200 mg tildrakizumab 59.6 ± 5.6% p ¼ 0.0003, 12 times weekly 200 mg tildrakizumab 66.6 ± 5.4% p < 0.0001, placebo 30.4 ± 5.2%) and MDA (4 times weekly 200 mg tildrakizumab 33.3 ± 5.3% p < 0.0001, 12 times weekly 200 mg til- drakizumab 34.2 ± 5.3% p < 0.0001, placebo 6.3 ± 2.7%) [51]. Improvements in skin were also seen with all doses of tildrakizumab vs. placebo at week 24, as measured by PASI100 (4 times weekly 200 mg tildrakizumab 30.2%, 12 times weekly 200 mg tildrakizumab 25.0%, 12 times weekly 100 mg tildrakizumab 273% and 12 times weekly 20 mg tildrakizumab 22.0%, placebo 4.8%) and sustained to week 52 [51]. No significant improvements in enthesitis and dactylitis were seen [51]. Tildrakizumab was generally well tolerated, with no significant reports of IBD, major adverse cardiac events or death through week 52 [51]. Phase III results for PsA are expected in Q2 of 2023 [52,53]. Of note, RCTs in plaque psoriasis have demonstrated significant and sustained efficacy, and tildrakizumab is licensed for plaque psoriasis by the EMA and FDA [54,55].
JAK and TYK2 inhibitors
Cytokines involved in the pathogenesis of PsA induce cellular activation, in part, through the JAK- STAT intracellular kinase signalling system (Fig. 2) [2]. This system comprises four key molecules; JAK1, JAK2, JAK3 and TYK2, with inhibitors targeting different members of this family with variable selectivity [2,56]. Tofacitinib was the first approved tsDMARD, and it showed selectivity for JAK1 and
Deucravacitinib
TYK2 is another intracellular kinase family member that mediates interferon (IFN)-a, IFN-b and
IL-12/23 signalling (Fig. 2) [64]. Deucravacitinib is an oral selective TYK2 inhibitor that binds to the regulatory domain of TYK2 with a high degree of selectivity [64]. This contrasts with other JAK inhibitory small molecules that bind to the conserved active domains of JAK1, 2 and 3 [64]. Phase II studies compared 6 mg vs. 13 mg deucravacitinib once daily vs. placebo in 203 patients with active PsA (csDMARD-IR and TNFi-IR in 15.8%) [64]. Both doses of deucravacitinib were statistically more likely than placebo at week 16 to achieve the primary endpoint of ACR20 (6 mg OR 2.4, 95% CI 1.2e4.8, p ¼ 0.0134; 12 mg OR 3.6, 95% CI 1.8e7.4, p ¼ 0.0004). Prior TNFi exposure did not appear to influence these results. Deucravacitinib treatment was associated with statistically greater im- provements in all secondary endpoints including musculoskeletal disease (ACR50 and ACR70), skin (PASI75), enthesitis, dactylitis and disability (HAQ-DI) compared to placebo [64]. Both doses were associated with higher frequencies of adverse events, including nasopharyngitis, URTI, sinusitis, bronchitis, rashes, diarrhoea and headaches. No significant opportunistic infections, malignancies or thromboembolic events were noted in treated patients [64]. Phase III studies are ongoing for PsA [65]. Superiority of deucravacitinib compared to both placebo and apremilast in moderate-to-severe plaque psoriasis has also been shown [66,67]. Based on these phase 3 trials, deucravacitinib is now approved for the treatment of psoriasis by the FDA and other regulatory agencies. The safety label is different from other JAK inhibitors in the following ways: 1) there is no black box warning regarding the risk of major cardiovascular events, thromboembolic events, malignancy or death; 2) prescrip- tion of deucravacitinib does not have to be preceded by a trial of a TNF inhibitor; and 3) laboratory monitoring is only required in patients with hepatotoxic risk; otherwise, routine laboratory moni- toring is not required.
Brepocitinib Brepocitinib is a small molecule that inhibits both JAK1 and TYK2 (Fig. 2) [68]. A phase IIB study compared 10 mg, 30 mg and 60 mg once daily brepocitinib vs. placebo in 218 patients with active PsA (csDMARD-IR and TNFi-IR in up to 30%). Significantly higher proportions of patients treated with 30 mg and 60 mg brepocitinib achieved the primary outcome of ACR20 at week 16 compared to placebo (30 mg 40%, 60 mg 44%, placebo 29%). Both 30 mg and 60 mg brepocitinib doses were associated with significantly higher response rates when compared to placebo at 16 weeks for secondary endpoints in musculoskeletal (ACR50, ACR70 and MDA), skin (PASI75 and PASI90), functioning (HAQ-DI) and fatigue (FACIT-Fatigue) domains [68]. The higher brepocitinib dose of 60 mg was statistically more likely than placebo to achieve secondary endpoints in enthesitis and dactylitis at week 16 [68]. These improve- ments were maintained for 52 weeks [68]. There were more adverse events in the 30 mg and 60 mg brepocitinib treatment groups than in the placebo over the 16-week treatment period (30 mg 55%, 60 mg 66.7%, placebo 47.8%). Most were mild and in keeping with other JAKi, such as herpes zoster infection and blood test derangements. No major safety signals have been reported to date [68]. A phase 3 trial in PsA is not yet planned; however, trials in dermatomyositis and non-infectious uveitis are in process.
Emerging therapeutics for PsA
Mitogen-activated protein kinase 2 (MK2) inhibitors
The p38 mitogen-activated protein kinase (MAPK) pathway is a crucial controller of stress-
mediated production of a variety of pro-inflammatory cytokines including TNF, IFNg, IL-1b, IL-6,
IL-12 and IL-17 [69e71]. One such downstream target of p38-MAPK is MK2, which acts to increase the mRNA stability and translation of pro-inflammatory genes. This is through the phosphorylation of a zinc-binding protein tristetraprolin (TTP) that, in its unphosphorylated form, binds to AU-rich areas in untranslated regions of mRNA and destabilises them [71]. Phosphorylation of TTP reduces its affinity, allowing the RNA-stabilising protein Human antigen R (HuR) to bind, thereby promoting
translation (Fig. 3) [71]. Inhibition of p38-MAPK has not been successful to date due to a combination of factors including cardiotoxicity, hepatotoxicity and inefficacy [69,71e73]. A notable phenomenon has been the progressive decrease in response after repeat administrations (tachyphylaxis) and is believed to be related to the unwanted pleiotropic effects of p38-MAPK inhibition [71]. Therefore, downstream proteins including MK2 have been identified as potential therapeutic targets and have begun preliminary assessment.
Fig. 3. Novel therapeutic targets for PsA. Figure summarises the mechanism of action of novel and/or in-pipeline medications for PsA. Briefly, IL-17A and IL-17F are produced by cells including Th17, gd T-cells, ILCs, iNK T-cells, MAIT cells, neutrophils and mast cells as both homo- and hetero-dimers. This binds to the IL-17 receptor consisting of 2 subunits, IL-17RA and IL-17RC, recruiting the ACT adaptor, which recruits the downstream effectors TRAF2, TRAF5 and TRAF6, which in turn activate the NF-kb pathway. Izokibep inhibits IL-17A. Sonelokimab inhibits IL-17A, IL-17F and IL-17 heterodimers. Brodalumab inhibits IL-17RA. IL-23 and TNF are pro- duced from activated DCs and macrophages, as well as other immune and stromal cells. GM-CSF is produced mainly by DCs and macrophages, with the net result of pro-inflammatory cytokine production. Otilimab inhibits GM-CSF. ABBV-3773 is a glucocorticoid receptor modulator combined with TNFi that works on cells with membrane-bound TNF. Finally, the p38-MAPK pathway is activated by noxious stimuli including TNF, IL-1B, IL-8 and LPS. These activate MKK3 and MKK6, which in turn recruit p38 kinase. This targets MK2, which acts to increase the mRNA stability and translation of pro-inflammatory genes. PsA, psoriatic arthritis; IL, interleukin; ILCs, innate-like lymphocytes; iNK, induced natural killer; MAIT, mucosal associated invariant T; IL-17RA, interleukin-17 receptor subunit A; IL-17RC, interleukin-17 receptor subunit C; TRAF, tumour necrosis factor receptor associated factor; NF-kb, nuclear factor kappa beta; GM-CSF, granulocyte macrophage colony stimulating factor; TNFi, tumour necrosis factor inhibitor; TNF, tumour ne- crosis factor; MAPK, mitogen-activated protein kinase; LPS, lipopolysaccharide; MKK, mitogen activated protein kinase kinase; MK2, mitogen-activated protein kinase-activated protein kinase 2.
A phase IIB multicentre randomised trial assessed sonelokimab 30 mg vs. 60 mg vs. 120 mg normal loading dose vs. sonelokimab 120 mg augmented loading dose vs. 300 mg secukinumab comparator vs. placebo in 313 patients with moderate-severe plaque psoriasis (csDMARDs-IR, �2 bDMARDs-IR; but prior IL-17i experienced patients were excluded) [80]. Participants underwent a 12-week placebo-controlled induction, 12-week dose maintenance or escalation period, followed by a 24- week response assessment period. All doses of sonelokimab were statistically more likely than placebo at week 12 to achieve the primary endpoint of investigator's global assessment (IGA) score of 0 e1 (placebo 0%, 95% CI 0e6.8%; 30 mg 48.1%, 95% CI 34e62.4%, p < 0.0001; 60 mg 84.6%, 95% CI 71.9e93.1%, p < 0.0001; 120 mg normal load 77.4%, 95% CI 63.8e87.7%, p < 0.0001; 120 mg augmented load 88.2%, 95% CI 76.1e95.6%, p < 0.0001; 300 mg 77.4%, 95% CI 63.8e87.7%, p < 0.0001) [80]. Doses of sonelokimab 120 mg with augmented load were significantly more likely than placebo at week 12 to achieve skin responses of PASI90 (76.5%, 95% CI 62.5e87.2%, p < 0.0001) and PASI (33.3%, 95% CI 20.8e47.9%, p < 0.0001) [80]. During the dose maintenance and escalation periods, patients previously treated with placebo had rapid improvements in their PASI scores when tran- sitioned to sonelokimab [80]. Mild-to-moderate adverse events were reported, including naso- pharyngitis, pruritis, URTI, and notably, one patient with de novo Crohn's disease. During the 52- week trial, the overall safety profile was similar to secukinumab, with the exception of a possible minor increase in Candida infections [80]. Serious Candida infections were similar between secuki- numab and sonelokimab (N ¼ 1 in both arms). There are no registered RCTs for PsA on clinicaltrials. gov at the time of writing.
GM-CSF inhibitors
GM-CSF is a key driver of inflammation, resultant pain and tissue damage in a range of IMIDs, including PsA [81,82]. GM-CSF is produced by fibroblasts, endothelial cells and CD163þ sublining macrophages [81]. GM-CSF can enhance the activation of myeloid cells and the production of pro- inflammatory mediators [82]. Murine studies have associated GM-CSF with the development of pain behaviour, potentially through the over-sensitisation of sensory nerves [82]. Otilimab (also known as GSK3196165, MOR103 and MOR04357) is a high affinity recombinant human IgG1 monoclonal anti- body antagonist of GM-CSF [82]. The phase IIb BAROQUE study assessed otilimab 22.5 mg vs. 45 mg vs. 90 mg vs. 135 mg vs. 180 mg vs. placebo, with concomitant methotrexate, in 222 patients with moderate-to-severe RA (csDMARDs- IR) [82]. Treatments were given weekly for 5 weeks, then on alternate weeks until week 50. Patients without a good-moderate EULAR response at week 12 or a DAS28-CRP<3.2 at week 24 were escalated to otilimab 180 mg. The primary endpoint of DAS28-CRP <2.6 at week 24 was not met for any treat- ment group, though the DAS28-CRP <2.6 remission rates were numerically higher for all otilimab treatment groups when compared to placebo [82]. The largest difference, although not statistically significant, was seen with otilimab 90 mg (OR 8.39, 95% CI 0.98e72.14, p ¼ 0.053) [82]. Post-hoc analysis indicated that the proportion of patients achieving DAS28-CRP <2.6 increased with increasing otilimab dose. Significant improvements in DAS28-CRP <2.6 change from baseline were seen with otilimab 180 mg compared to placebo at both 12 weeks (difference �1.27, 95% CI -1.91 to 0.63, p ¼ 0.0001) and 24 weeks (difference �1.82, 95% CI -2.75 to �0.89, p ¼ 0.002) [82]. Significantly more patients receiving any otilimab dose achieved the secondary musculoskeletal endpoints of ACR20 (22.5 mg 35%, 45 mg 41%, 90 mg 51%, 135 mg 41%, 180 mg 51%, placebo 11%; p < 0.05), ACR50 for the 45 mg and 135 mg doses (45 mg 27%, 135 mg 30%, placebo 8%) and good/moderate EULAR responses particularly with the 180 mg dose (difference vs. placebo 54.1%, 95% CI 34.9e73.2, p < 0.0001) [82]. Improvements in patient reported outcomes, including FACIT-Fatigue (difference vs. placebo 8.7%, p < 0.05), pain (difference vs. placebo �25.01, p < 0.001) and patient global assessment (difference vs. placebo �23.90, p < 0.001) were also significantly more likely with the otilimab 180 mg [82]. Adverse events were equivalent across treatment groups, and most were mild-moderate. Common side effects included nasophar- yngitis, URTI and anaemia [82]. The phase III ContRAst RCT of RA patients has completed [83e85], and preliminary results are awaited. The long-term extension ContRAst-X arm continues to collect data [86]. No studies for PsA are yet planned.
Other dual target therapies
Dual TNF inhibitor and glucocorticoid receptor modulators ABBV-3373 is a dual TNFi (adalimumab) with a conjugated glucocorticoid receptor modulator [87]. ABBV-3373 aims to deliver glucocorticoids directly to activated leukocytes expressing membrane- bound TNF (Fig. 3). It is hoped that this will provide significant anti-inflammatory effects whilst minimising systemic glucocorticoid side effects. A phase IIa double-blinded randomised controlled trial compared ABBV-3773 100 mg for 12 weeks followed by placebo for 12 weeks vs. fortnightly adalimumab 80 mg for 48 weeks in 48 patients with moderate-severe RA (csDMARDs-IR) [87]. Adalimumab data was also supplemented with pre-specified historical adalimumab outcome data. ABBV-3773 treatment was statistically more likely than pre- specified historical adalimumab at 12 weeks to achieve the primary endpoint of improvement in DAS28-CRP (ABBV-3773 -2.65 vs. adalimumab �2.13, p ¼ 0.022) and not statistically different to combined trial and historical adalimumab data (ABBV-3773 -2.65 vs. adalimumab �2.29, probability 90%) [87]. Comparable improvements in secondary endpoints such as disease activity as measured by the clinical disease activity index (CDAI), simplified disease activity index (SDAI), ACR20, ACR50 and ACR70 were seen with ABBV-3773 and in-trial adalimumab [87]. No systemic glucocorticoid effects (serum cortisol level change) were noted. ABBV-3373 had a similar safety profile to adalimumab. Two serious infections (lower respiratory tract infection and URTI) and one case of anaphylaxis were re- ported in ABBV-3373 treated patients [87]. No studies have yet been reported or registered on its ef- ficacy or safety in PsA.
Dual TNF and IL-17 inhibitors ABT-122 is a dual variable domain immunoglobulin that inhibits both TNF and IL-17A with high affinity [88]. ABT-122 is built on the backbone of adalimumab with added IL-17A binding domains [88]. Targeting both molecules is hoped to achieve better responses than targeting either molecule alone. A phase II trial compared ABT-122 120 mg vs. 240 mg with placebo (including an active comparator arm of 40 mg adalimumab) in 240 patients with active PsA (csDMARDs-IR) [88]. Both 120 mg and 240 mg of ABT-122 were statistically more likely than placebo at week 12 to achieve the primary endpoint of ACR20 (120 mg 64.8%, 250 mg 75.3%, placebo 25% response rate, p < 0.001 and comparable to those receiving adalimumab (68.1% response rate) [88]. Both doses of ABT-122 were statistically more likely than placebo to achieve the secondary endpoints at week 12 for musculoskeletal disease including ACR50 (ABT-122 240 mg 53.4%, ABT-122 120 mg 36.6%, placebo 12.5% p < 0.001) and ACR70 (ABT- 122 120 mg 22.5%, ABT-122 240 mg 31.5%, placebo 4.2, p ¼ 0.034 and p ¼ 0.004, respectively) [88]. The higher 240 mg dose of ABT-122 was also statistically more likely than adalimumab to achieve ACR (ABT-122 240 mg 53.4%, adalimumab 37.5%, p ¼ 0.039), ACR70 (ABT-122 240 mg 31.5%, adalimumab 15,3%, p ¼ 0.017) and mean changes in DAS28-CRP (ABT-122 -2.28, adalimumab �1.83, p ¼ 0.012) [88]. Similar significant improvements were seen in SPARCC enthesitis index (ABT-122 120 mg �2.8, ABT- 122 240 mg �2.5, placebo �1.2, p ¼ 0.003 and 0.017, respectively) [88]. Both ABT-122 120 mg and 240 mg were statistically more likely to achieve skin responses at week 12 when compared to placebo as measured by PASI75 (ABT-122 120 mg 74.4%, ABT-122 240 mg 77.6%, placebo 27%, p < 0.01) [88]. Again, the 240 mg dose of ABT-122 was statistically more likely than adalimumab to achieve a PASI75 at week 12 (ABT-122 240 mg 77.6%, adalimumab 57.6%, p ¼ 0.047) [88]. Adverse effects including infection incidence were similar across patient groups, though two patients in the ABT-122 groups had non-serious oral candidiasis, and one patient receiving 240 mg ABT-122 developed sick sinus syn- drome [88]. ABT-122 has also undergone phase II trials in RA, with similar improvements seen in ACR20, ACR50 and ACR70, though no superiority to adalimumab was observed [89]. At present, no further trials in PsA are planned.
Dual TNF and IL-23 inhibitors Dual inhibitors of both TNF and IL-17 on the same agent have been proposed. In the Mycobacterium
tuberculosis HLA-B27/b2-microglobulin transgenic rat models of spondylarthritis (SpA), dual blockade
reduced clinical spondylitis, peripheral arthritis both clinically and radiographically via micro-CT [90]. Dual blockade also reduced pannus formation and inflammation evident on histology [90]. The phase
Research Agenda
� Cellular and molecular phenotyping of disease-relevant tissues in patients at different disease stages should improve our understanding of the pathophysiology of PsA. Integrating this with biomarkers and imaging might allow us to prevent the progression of psoriasis to PsA, apply pre- cision and stratified medicine approaches, and identify new therapeutic targets. � The application of emerging artificial intelligence methodologies in big data sets could be used to better identify therapeutic targets, design novel trials that better address the different phenotypes (subsets) of PsA that have been neglected to date, and better interprete the complex datasets produced by trials of heterogenous diseases such as PsA. � Prospective pragmatic trials are then needed to assess the long-term efficacy, safety and health economics of emerging therapeutics in clinical practice, beyond the selective and controlled en- vironments of RCTs.
Declaration of competing interest
D.J. declares that he has received research grants, education grants and/or honoraria from phar- maceutical companies, including AbbVie, Amgen, Biogen, Celgene, Eli Lilly, Fresenius Kabi, Galapagos/ Gilead, GSK, Healthcare Celltrion, Janssen, Merck, Novartis, Pfizer, Roche, Sandoz and UCB. PM declares that he has received research grants, consultation fees, and/or speaker honoraria: AbbVie, ACELYRIN, Aclaris, Amgen, Bristol Myers, Boehringer-Ingelheim, Galapagos, Gilead, GlaxoSmithKline, Inmagene, Janssen, Lilly, MoonLake, Novartis, Pfizer, SUN Pharma, UCB JPH has nothing further to declare.
Acknowledgements
D.J. acknowledges that his research was supported by the Cambridge Arthritis Research Endeavour (CARE) and the NIHR Cambridge Biomedical Research Centre (BRC-1215-20014). JPH acknowledges that his research is supported by the Medical Research Council UK and the Evelyn Trust. [The views expressed are those of the authors and not necessarily those of the MRC, NIHR, Evelyn Trust or the Department of Health and Social Care]. The other authors received no specific grant from any funding agency in the public, commercial or not-for-profit sectors for the preparation of this manuscript.
References
[1] Veale DJ, Fearon U. The pathogenesis of psoriatic arthritis. Lancet 2018;391:2273e84. https://doi.org/10.1016/S0140- 6736(18)30830-4. [2] FitzGerald O, Ogdie A, Chandran V, et al. Psoriatic arthritis. Nat Rev Dis Prim 2021;7:59. https://doi.org/10.1038/s41572- 021-00293-y. [3] Shah K, Paris M, Mellars L, et al. Real-world burden of comorbidities in US patients with psoriatic arthritis. RMD Open 2017;3:e000588. https://doi.org/10.1136/rmdopen-2017-000588. [4] Tillett W, de-Vries C, McHugh NJ. Work disability in psoriatic arthritis: a systematic review. Rheumatology 2012;51: 275 e83. https://doi.org/10.1093/rheumatology/ker216. [5] Merola JF, Dennis N, Chakravarty SD, et al. Healthcare utilization and costs among patients with psoriasis and psoriatic arthritis in the USA-a retrospective study of claims data from 2009 to 2020. Clin Rheumatol 2021;40:4061e70. https:// doi.org/10.1007/s10067-021-05713-8. [6] Armstrong AW, Mehta MD, Schupp CW, et al. Psoriasis prevalence in adults in the United States. JAMA Dermatol 2021; 157:940e6. https://doi.org/10.1001/jamadermatol.2021.2007. [7] Coates LC, Moverley AR, McParland L, et al. Effect of tight control of inflammation in early psoriatic arthritis (TICOPA): a UK multicentre, open-label, randomised controlled trial. Lancet 2015;386:2489e98. https://doi.org/10.1016/S0140- 6736(15)00347-5. [8] Rombach I, Tillett W, Jadon D, et al. Treating to target in psoriatic arthritis: assessing real-world outcomes and optimising therapeutic strategy for adults with psoriatic arthritis-study protocol for the Monitor-PSA study, a trials within cohorts study design. Trials 2021;22:185. https://doi.org/10.1186/s13063-021-05142-7. [9] Tillett W, Jadon D, Shaddick G, et al. Smoking and delay to diagnosis are associated with poorer functional outcome in psoriatic arthritis. Ann Rheum Dis 2013;72:1358e61. https://doi.org/10.1136/annrheumdis-2012-202608. [10] Ng BCK, Jadon DR. Unmet needs in psoriatic arthritis. Best Pract Res Clin Rheumatol 2021;35:101693. https://doi.org/10. 1016/j.berh.2021.101693.
[11] van den Bosch F, Coates L. Clinical management of psoriatic arthritis. Lancet 2018;391:2285e94. https://doi.org/10.1016/ S0140-6736(18)30949-8. [12] Kane D, Stafford L, Bresnihan B, FitzGerald O. A prospective, clinical and radiological study of early psoriatic arthritis: an early synovitis clinic experience. Rheumatology 2003;42:1460e8. https://doi.org/10.1093/rheumatology/keg384. [13] Alten R, Conaghan PG, Strand V, et al. Unmet needs in psoriatic arthritis patients receiving immunomodulatory therapy: results from a large multinational real-world study. Clin Rheumatol 2019;38:1615e26. https://doi.org/10.1007/s10067- 019-04446-z. [14] Coates LC, Soriano ER, Corp N, et al. Group for research and assessment of psoriasis and psoriatic arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol 2022;18:465e79. https://doi.org/10. 1038/s41584-022-00798-0. [15] Mahmood F, Coates LC, Helliwell PS. Current concepts and unmet needs in psoriatic arthritis. Clin Rheumatol 2018;37: 297 e305. https://doi.org/10.1007/s10067-017-3908-y. [16] Kavanaugh A, Helliwell P, Ritchlin CT. Psoriatic arthritis and burden of disease: patient perspectives from the population- based multinational assessment of psoriasis and psoriatic arthritis (MAPP) survey. Rheumatol Ther 2016;3:91e102. https://doi.org/10.1007/s40744-016-0029-z. [17] Haroon M, Gallagher P, Heffernan E, FitzGerald O. High prevalence of metabolic syndrome and of insulin resistance in psoriatic arthritis is associated with the severity of underlying disease. J Rheumatol 2014;41:1357e65. https://doi.org/10. 3899/jrheum.140021. [18] Singh S, Facciorusso A, Singh AG, et al. Obesity and response to anti-tumor necrosis factor-a agents in patients with select immune-mediated inflammatory diseases: a systematic review and meta-analysis. PLoS One 2018;13:1e26. https://doi. org/10.1371/journal.pone.0195123. [19] Eder L, Thavaneswaran A, Chandran V, et al. Obesity is associated with a lower probability of achieving sustained minimal disease activity state among patients with psoriatic arthritis. Ann Rheum Dis 2015;74:813e7. https://doi.org/10.1136/ annrheumdis-2013-204448. [20] Intercollegiate S, Network G. Management of obesity. 2010 (SIGN Guideline No 115). [21] Aveyard P, Lewis A, Tearne S, et al. Screening and brief intervention for obesity in primary care: a parallel, two-arm, randomised trial. Lancet 2016;388:2492e500. https://doi.org/10.1016/S0140-6736(16)31893-1. [22] Wilding JPH, Batterham RL, Calanna S, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med 2021;384:989e1002. https://doi.org/10.1056/NEJMoa2032183. [23] Maglio C, Peltonen M, Rudin A, Carlsson LMS. Bariatric surgery and the incidence of psoriasis and psoriatic arthritis in the Swedish obese subjects study. Obesity 2017;25:2068e73. https://doi.org/10.1002/oby.21955. [24] Neovius M, Narbro K, Keating C, et al. Health care use during 20 years following bariatric surgery. JAMA 2012;308: 1132 e41. https://doi.org/10.1001/2012.jama.11792. [25] Elalouf O, Chandran V. Novel therapeutics in psoriatic arthritis. What is in the pipeline? Curr Rheumatol Rep 2018;20. https://doi.org/10.1007/s11926-018-0746-0. [26] Tahir H, Grewal S. Current unmet needs and emerging novel pharmacotherapies in psoriatic arthritis. Expet Opin Pharmacother 2022;23:417e20. https://doi.org/10.1080/14656566.2021.2006184. [27] Stober C, Ye W, Guruparan T, et al. Prevalence and predictors of tumour necrosis factor inhibitor persistence in psoriatic arthritis. Rheumatology 2018;57:158e63. https://doi.org/10.1093/rheumatology/kex387. [28] Egeberg A, Rosenø NAL, Aagaard D, et al. Drug survival of biologics and novel immunomodulators for rheumatoid arthritis, axial spondyloarthritis, psoriatic arthritis, and psoriasis - a nationwide cohort study from the DANBIO and DERMBIO registries. Semin Arthritis Rheum 2022;53:151979. https://doi.org/10.1016/j.semarthrit.2022.151979. [29] Balsa A, Lula S, Marshall L, et al. The comparative immunogenicity of biologic therapy and its clinical relevance in psoriatic arthritis: a systematic review of the literature. Expet Opin Biol Ther 2018;18:575e84. https://doi.org/10.1080/ 14712598.2018.1450385. [30] Mease PJ, Gladman DD, Collier DH, et al. Etanercept and methotrexate as monotherapy or in combination for psoriatic arthritis: primary results from a randomized, controlled phase III trial. Arthritis Rheumatol 2019;71:1112e24. https://doi. org/10.1002/art.40851. [31] Ariani A, Santilli D, Mozzani F, et al. Cycling or swap biologics and small molecules in psoriatic arthritis: observations from a real-life single center cohort. Medicine 2021:100. [32] Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol 2022. https://doi.org/10. 1038/s41577-022-00746-9. [33] Li X, Bechara R, Zhao J, et al. IL-17 receptorebased signaling and implications for disease. Nat Immunol 2019;20: 1594 e602. https://doi.org/10.1038/s41590-019-0514-y. [34] Mease PJ, Smolen JS, Behrens F, et al. A head-to-head comparison of the efficacy and safety of ixekizumab and adali- mumab in biological-na{"\i}ve patients with active psoriatic arthritis: 24-week results of a randomised, open-label, blinded-assessor trial. Ann Rheum Dis 2020;79:123e31. https://doi.org/10.1136/annrheumdis-2019-215386. [35] McInnes IB, Behrens F, Mease PJ, et al. Secukinumab versus adalimumab for treatment of active psoriatic arthritis (EXCEED): a double-blind, parallel-group, randomised, active-controlled, phase 3b trial. Lancet 2020;395:1496e505. https://doi.org/10.1016/S0140-6736(20)30564-X. [36] Gottlieb AB, Merola JF, Reich K, et al. Efficacy of secukinumab and adalimumab in patients with psoriatic arthritis and concomitant moderate-to-severe plaque psoriasis: results from EXCEED, a randomized, double-blind head-to-head monotherapy study. Br J Dermatol 2021;185:1124e34. https://doi.org/10.1111/bjd.20413. [37] Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 2018;55:379e90. https://doi.org/10.1007/s12016-018-8702-3. [38] Ritchlin CT, Kavanaugh A, Merola JF, et al. Bimekizumab in patients with active psoriatic arthritis: results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet 2020;395:427e40. https://doi.org/10. 1016/S0140-6736(19)33161-7. [39] Deodhar A, Gossec L, Mease P, et al. Bimekizumab treatment is associated with improvements in back pain and fatigue in patients with active psoriatic arthritis: 48-week results from a phase 2b study. Arthritis Rheumatol 2020:72.
TNFa Inhibitor Treatmen. Bethesda (MD): US National Library of Medicine n.d. https://clinicaltrials.gov/ct2/show/ NCT04908189 (accessed September 15, 2022). [66] Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled phase 3 POETYK PSO-1 trial. J Am Acad Dermatol 2022. https://doi.org/10.1016/j.jaad.2022.07.002. [67] Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, phase 3 POETYK PSO-2 trial. J Am Acad Dermatol 2022. https://doi.org/10.1016/j.jaad.2022.08.061. [68] Mease P, Helliwell P, Silwinska-Stanczyk P, et al. Efficacy and safety of brepocitinib (tyrosine kinase 2/janus kinase 1 inhibitor) for the treatment of active psoriatic arthritis: results from a phase 2b randomized controlled trial. Arthritis Rheumatol 2021;73. [69] Singh RK, Diwan M, Dastidar SG, Najmi AK. Differential effect of p38 and MK2 kinase inhibitors on the inflammatory and toxicity biomarkers in vitro. Hum Exp Toxicol 2018;37:521e31. https://doi.org/10.1177/0960327117715901. [70] Ramírez-Valle F, Adams M, Beebe L, et al. A novel MK2 inhibitor for the treatment of ankylosing spondylitis and other inflammatory diseases. Arthritis Rheumatol 2019;71. [71] Gaur R, Mensah KA, Stricker J, et al. CC-99677, a novel, oral, selective covalent MK2 inhibitor, sustainably reduces pro- inflammatory cytokine production. Arthritis Res Ther 2022;24:199. https://doi.org/10.1186/s13075-022-02850-6. [72] Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum 2009;60:1232e41. https://doi.org/10.1002/art.24485. [73] Dulos J, Wijnands FPG, van den Hurk-van Alebeek JAJ, et al. p38 inhibition and not MK2 inhibition enhances the secretion of chemokines from TNF-alpha activated rheumatoid arthritis fibroblast-like synoviocytes. Clin Exp Rheumatol 2013;31: 515 e25. [74] Gordon D, Hellriegel ET, Hope HR, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the MK2 in- hibitor ATI-450 in healthy subjects: a placebo-controlled, randomized phase 1 study. Clin Pharmacol 2021;13:123e34. https://doi.org/10.2147/CPAA.S305308. [75] Study of ATI-450 vs placebo in patients with moderate to severe psoriatic arthritis. Bethesda (MD): US National Library of Medicine; 2022. https://clinicaltrials.gov/ct2/show/NCT05511519 (accessed September 15, 2022). [76] A study of CC-99677 in participants with active ankylosing spondylitis (AS SpA axSpA). Bethesda (MD): US National Library of Medicine; 2021. https://clinicaltrials.gov/ct2/show/NCT04947579 (accessed September 15, 2022). [77] Behrens F, Taylor PC, Wetzel D, et al. OP0258 izokibep (ABY-035) in patients with active psoriatic arthritis e 16-WEEK results from a phase 2 study. Ann Rheum Dis 2022;81:170. https://doi.org/10.1136/annrheumdis-2022-eular.536. LP e
[78] Affibody. Affibody, ACELYRIN, and Inmagene biopharmaceuticals announce data from global phase 2 trial of izokibep in patients with psoriatic arthritis presented during 2022 European alliance of associations for rheumatology congress;
- https://www.affibody.se/affibody-acelyrin-and-inmagene-biopharmaceuticals-announce-data-from-global-phase- 2-trial-of-izokibep-in-patients-with-psoriatic-arthritis-presented-during-2022-european-alliance-of-associations-for/. [Accessed 15 September 2022]. accessed. [79] Acelyrin Acelyrin, Inc. Announces $300 million series C financing to accelerate phase 3 development of izokibep, a unique IL-17a inhibitor to treat inflammatory diseases. 2022. https://www.acelyrin.com/news/acelyrin-announces-300- million-series-c-financing (accessed September 15, 2022). [80] Papp KA, Weinberg MA, Morris A, Reich K. IL17A/F nanobody sonelokimab in patients with plaque psoriasis: a multi- centre, randomised, placebo-controlled, phase 2b study. Lancet 2021;397:1564e75. https://doi.org/10.1016/S0140- 6736(21)00440-2. [81] Fuentelsaz-Romero S, Cuervo A, Estrada-Capetillo L, et al. GM-CSF expression and macrophage polarization in joints of undifferentiated arthritis patients evolving to rheumatoid arthritis or psoriatic arthritis. Front Immunol 2020;11:613975. https://doi.org/10.3389/fimmu.2020.613975. [82] Buckley CD, Simon-Campos JA, Zhdan V, et al. Ef� ficacy, patient-reported outcomes, and safety of the anti-granulocyte macrophage colony-stimulating factor antibody otilimab (GSK3196165) in patients with rheumatoid arthritis: a rando- mised, phase 2b, dose-ranging study. Lancet Rheumatol 2020;2:e677e88. https://doi.org/10.1016/S2665-9913(20)30229-
[83] Efficacy and safety of GSK3196165 versus placebo and tofacitinib in participants with moderately to severely active rheumatoid arthritis who have an inadequate response to methotrexate (contRAst 1). Bethesda (MD): US National Li- brary of Medicine; 2022. https://clinicaltrials.gov/ct2/show/NCT03980483?term¼otilimab&draw¼ 2 &rank¼6 (accessed September 15, 2022). [84] Efficacy and safety of GSK3196165 versus placebo and tofacitinib in participants with moderately to severely active rheumatoid arthritis who have an inadequate response to conventional synthetic (cs)/Biologic (b) disease modifying anti-rheumatic drugs. DM. Bethesda (MD): US National Library of Medicine; 2022. https://clinicaltrials.gov/ct2/show/ NCT03970837?term¼otilimab&draw¼ 2 &rank¼5 (accessed September 15, 2022). [85] Efficacy and safety of GSK3196165 (otilimab) versus placebo and sarilumab in participants with moderately to severely active rheumatoid arthritis who have an inadequate response to biological disease-modifying antirheumatic drug (DMARDs) and/or janus kina. Bethesda (MD): US National Library of Medicine; 2022. https://clinicaltrials.gov/ct2/show/ NCT04134728?term¼otilimab&draw¼ 2 &rank¼4 (accessed September 15, 2022). [86] Long-term safety and efficacy of GSK3196165 (otilimab) in the treatment of rheumatoid arthritis (RA) (contRAst X). Bethesda (MD): US National Library of Medicine; 2022. https://clinicaltrials.gov/ct2/show/NCT04333147 (accessed September 15, 2022). [87] Buttgereit F, Aelion J, Rojkovich B, et al. OP0115 efficacy and safety of ABBV-3373, a novel anti-tnf glucocorticoid receptor modulator antibody drug conjugate, in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy: a phase 2A proof of concept study. Ann Rheum Dis 2021;80:64. https://doi.org/10.1136/annrheumdis-2021- eular.221. LP e 64.
[88] Mease PJ, Genovese MC, Weinblatt ME, et al. Phase II study of ABT-122, a tumor necrosis factore and interleukin- 17aetargeted dual variable domain immunoglobulin, in patients with psoriatic arthritis with an inadequate response to methotrexate. Arthritis Rheumatol 2018;70:1778e89. https://doi.org/10.1002/art.40579. [89] Genovese MC, Weinblatt ME, Aelion JA, et al. ABT-122, a bispecific dual variable domain immunoglobulin targeting tumor necrosis factor and interleukin-17a, in patients with rheumatoid arthritis with an inadequate response to methotrexate. Arthritis Rheumatol 2018;70:1710e20. https://doi.org/10.1002/art.40580. [90] Au - Bryant Hammoura Renee H, Au - Westmoreland Shaughn H, SusanAU - Kingsbury, et al. TI - dual blockad I-F. Dual blockade of TNF and IL-17a inhibits inflammation and structural damage in a rat model of spondyloarthritis. Int J Mol Sci 2022;23. https://doi.org/10.3390/ijms23020859. [91] A study of guselkumab and golimumab combination therapy in participants with active psoriatic arthritis (AFFINITY). Bethesda (MD): US National Library of Medicine; 2022. https://clinicaltrials.gov/ct2/show/NCT05071664 (accessed September 15, 2022). [92] Buckley CD, McGettrick HM. Leukocyte trafficking between stromal compartments: lessons from rheumatoid arthritis. Nat Rev Rheumatol 2018;14:476e87. https://doi.org/10.1038/s41584-018-0042-4. [93] Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheu- matol 2016;12:5e13. https://doi.org/10.1038/nrrheum.2015.157. [94] Guagnozzi D, Caprilli R. Natalizumab in the treatment of Crohn's disease. Biologics 2008;2:275e84. [95] Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med 2013;369:711e21. https://doi.org/10.1056/NEJMoa1215739. [96] Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2013;369:699e710. https://doi.org/10.1056/NEJMoa1215734. [97] García-Vicu~na R, Brown M. Vedolizumab for inflammatory bowel disease: a two-edge sword in the gut-joint/enthesis axis. Rheumatology 2019;58. https://doi.org/10.1093/rheumatology/key440. [98] Alivernini S, MacDonald L, Elmesmari A, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat Med 2020;26:1295e306. https://doi.org/10.1038/s41591-020-0939-8. [99] Croft AP, Campos J, Jansen K, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019; 570:246e51. https://doi.org/10.1038/s41586-019-1263-7. [100] Miyagawa I, Nakayamada S, Nakano K, et al. Precision medicine using different biological DMARDs based on charac- teristic phenotypes of peripheral T helper cells in psoriatic arthritis. Rheumatology 2019;58:336e44. https://doi.org/10. 1093/rheumatology/key069.